

次世代シーケンシング(Next Generation Sequencing, NGS)は、ランダムに切断された数千万―数億のDNA断片の塩基配列を同時並行的に決定する実験手法です。断片化した1本鎖のDNAに対して、断片の相補鎖を合成しながら決定する手法(SBS, Sequence-by-Synthesis法)によって、塩基配列を決定します。
次世代シーケンシングは大型の次世代シーケンサーの他、近年ではベンチトップサイズの機器、あるいはハンディタイプの機器などが現れています。それらのすべてについて、機器から出力された塩基配列情報を処理・解析するためのコンピューターが必要です。このため、バイオインフォマティクスについての知識が近年ますます重要となってきています。
ChIP-seqにおけるNGS出力データ分析
一般に、すべてのNGSデータ分析ワークフローは、1次、2次、3次分析に分けられますが、その定義はシーケンシング機器メーカーおよび受託NGS解析業者によって微妙に異なっています。
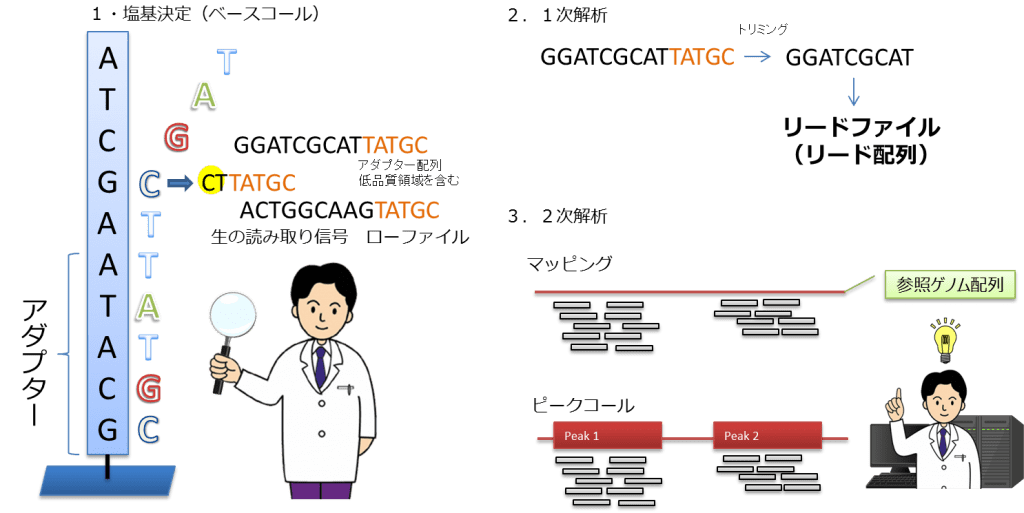
一次分析では、シーケンシングマシンからSBS法など各社技術により同時並行的に生成される生の読み取り信号のトリミング処理および品質のチェックを行います。
信号を塩基に変換してリード配列とします。一次分析が終了すると、すべてのリード配列を含むリードファイルが生成されます。
二次分析は、主にアラインメントから成り、ここでリード配列は参照ゲノムにマッピングされます。配列決定に加えて、リード配列のマップ上での集積情報を含むピークコールもその後に実行されます。
このように、二次解析によりアライメントファイルと検出されたピークおよび濃縮されたサンプルの部位情報が得られます。多くの受託業者はここまでの分析をシーケンシング業務として請け負うことが多いようです。
三次分析は、通常、研究者が特定の研究課題に対応する分析を行うため、「意味づけ」と呼ばれることがあります。従って、本質的に、三次分析法は多様で特異的であり、各研究ごとに異なる可能性があります。
ChIP-seqデータ解析ワークフロー
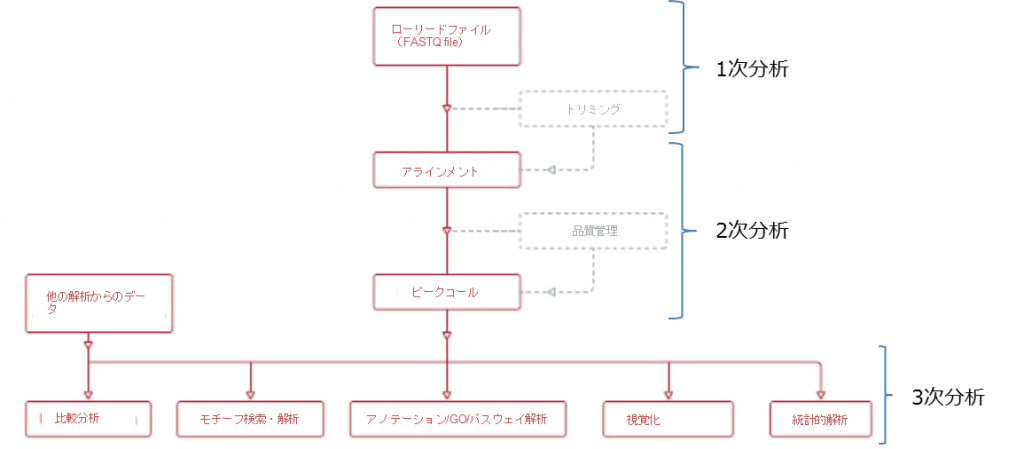
分析ワークフローの各フェーズには、データ処理とQCのさまざまなステップが含まれています。次のセクションでは、二次分析から、リードファイルの処理の手順を順を追って説明します。
1. アラインメント
リードをマッピングするには、通常はBWAが使用されますが、他のツールを使用することも可能です(たとえば、ELAND、TMAP、Bowtie)。 通常は豊富なリード量があるため、できるだけ多くのリード配列をアラインメントさせ、ミスアライメントを避けることが重要です。大部分のアラインメントソフトは1〜2のミスマッチしか許容しません。
2. リード配列の品質管理
FastQCを使用することによって、信頼性の高いリード配列の品質管理を行うことが可能です。一般的な配列の品質、読み取り長さ分布、GC含量、アダプター配列のコンタミネーション、重複読み取りレベルなどを報告するので非常に有益です。リード配列、またはアラインメントの両方で使用できます。
3. 相互相関分析
ENCODEのガイドラインによってCross-correlation profileとして提案されている分析法で、リード配列をゲノム上に配置した後得られるピークのS/N比を計測することが可能です。簡潔に手法を要約すると、この方法は、ゲノムに沿って順および逆鎖のリードをスライドさせます。すると理論的には、リード分布はスライドさせた「ずれ」がフラグメント長のサイズになったところで合致します(すなわち、リード分布はフラグメントの平均長さにほぼ等しい距離で相関します)。この相関の度合いを用いて、サンプルの濃縮レベルの評価をS/N比として行うことが可能です。
4. ピークコール
ピークコールとは、ゲノムからリード配列が有意に濃縮している箇所を網羅的に同定する作業です。
5. データセットとアノテーションの特徴付け
3次分析には、ChIP-seqデータセットの品質を制御し、相互に比較するために使用するカスタマイズされた手順が含まれます。また、遺伝子を可視化するため配列のアノテーションを行います。データの比較を行うための重要なパラメータが数多くありますが、これらの数字は、個々にではなく常にデータの意図に対し最適なものを選択する必要があります。
6. 重複分析
ChIP-seqデータセットの品質を評価する際には、上記のパラメータをチェックするだけでなく、2つのデータセットでピーク位置が一致するかどうかを判断することによって、参照データセットとの比較が最も重要です。これは、期待されるピークを再現することに成功したことを示しています。適切な参照データセットを選択することは、この分析の最も重要な部分です。

エピジェネティクス受託サービス
DiagenodeではChIP、RNA、DNAメチル化にわたる広範囲のエピゲノム解析について、蓄積してきた専門知識と信頼をベースに受託サービスを提供しています。リンク先では解析サービスの概要と各分析サービスについての説明へのリンクもご覧いただけます。
CATS RNA-seq Kit v2 x24
DiagenodeのCATS RNA-seq Kitは、少ないRNA量から次世代シーケンシングに使用するDNAライブラリーを作製できるライゲーションフリーのライブラリー調製キットです。リンク先ではキット概要などをご覧いただけます。
MicroPlex Library Preparation Kit v2 (12 indices)
MicroPlex Library Preparation™キットは、ChIP-seqで検証され、ピコグラムオーダーのDNAからライブラリーを作成できる唯一のキットです。製品の紹介と製品に関する研究者の声をご覧いただけます。
Premium Reduced Representation Bisulfite Sequencing (RRBS) kit
Reduced Representation Bisulfite Sequencing (RRBS)はCpG部位を含むDNAを濃縮することによって効率よく、単一ヌクレオチドレベルでDNAメチル化を分析することのできる手法です。キット製品紹介の他、実験手法についてのウェビナーをご覧いただけます。