
データ解析 (14)
古いプロトコルでは、pA-Tn5融合タンパク質に混入している大腸菌DNAをspike-in DNAとして用いることが推奨されていますが、DiagenodeのpA-Tn5は大腸菌DNAの汚染がないため、この方法は提案していません。
通常、サンプル間で等しい数のインプット細胞数があることを確認する方法をお勧めしています。
正規化は、データ解析時に読み取り深度(カバレッジ)の相対値を計算することによって得ることも可能です。各サンプルからの読み取り値は、100万人あたりのカウントに正規化されます。
Genome Biol 23, 144 (2022). https://doi.org/10.1186/s13059-022-02707-w
もし、免疫沈降で濃縮したいタンパク質がどこに結合するのかについての情報がない場合、ChIP-seqで得られるNGSのピーク情報や配列情報以外の確実なアウトプットはありません。
まず確認できることとしては、既知のターゲットに対する抗体をポジティブコントロールとし、プロトコールが正常に行われているかについて確認してみることができます。この場合、抗体がおのずと異なるため、目的のタンパク質については確認できません。
また、qPCRプライマーデザインを行うために、目的タンパク質の結合モチーフの情報があればそれを利用する方法もあります。
もし実験に使用しているサンプルや類似サンプルで一度でもChIP-seqのデータが取得できた場合は、その実験結果を基にqPCRのプライマーデザインを行い、その後の同じ実験系で濃縮率を反映するか確認していくことも可能です。
Diagenode ChIP-seq受託解析サービスでは、シーケンスによるChIP実験結果の検証を行っておりますので、qPCRプライマーについても確実に設計できます。
一つの方法としては、「測定限界外」のCt値を仮に「>40」として値に40を取ることです。
すなわち、Ct=40をPCRサイクルで増幅が得られないサンプルとして定義します。
この値に基づき、サンプルの測定値を比較Ct法などを用いて相対定量計算を行います。
ここから言えるのは、サンプル測定値 vs. IgGコントロールによる正規化は通常ChIP-qPCRでは必要ないということです。
ChIP-qPCRでは免疫沈降サンプルのCtはインプットによって正規化されるのがより実用的です。
IgGのqPCRシグナルは通常免疫沈降シグナルよりかなり低く、全く検出されない場合もあるため、Ctを用いた正規化の計算には不向きと言えます。
特異性の評価については、別にIgGをインプットによって正規化したデータを使用して示すのが適しています。
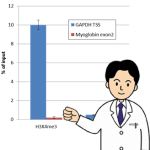
特に数量的データの正規化にはインプットからのリカバリー%をご利用ください。
IgGはバックグラウンドノイズがどの程度あるかについての指標にはなりますが、正規化に使用するにはインプットほど確度が高くありません。
qPCRの際は特にインプットを使用して正規化を行うことが重要です。
下のページについても併せてご覧ください。

ChIP-seqとRNA-seqの組み合わせによるデータ拡張は価値のあるデータセットを生成でき、マルチオミクス研究でも非常に多用されている組み合わせです。
多くのヒストンマークは遺伝子発現と関連することがわかっており、プロモーターやエンハンサーに対し活性化・poised・抑制化の3通りのコントロールを及ぼします。
特にヒストンマークについてRNA-seqのデータは貴重な追加の洞察を与えることができます。機能アノテーション分析(Functional annotation analysis)において、RNA-seqの情報を基に発現遺伝子のリストを微調整する目的にも使用できます。
解析を行う前に、ラボの担当者からデータ処理のパイプラインに何を使用したか、確認してください。論文では多くの場合何を使用したかは言及されていますが、重要なパラメータについて言及されてない場合もあるようです。
一般に、データの有効性を評価するためのいくつかのパラメータがあります。以下の観点からシーケンス品質を定量化します:
- ピークに含まれるリード数
- ピーク高さが高く、バックグラウンドが低い
- シーケンス深度
- 複雑性の高いライブラリー(重複が少ない)
- コントロールにおけるターゲットの濃縮が低い
- レプリケート間で値が相似する
- 遺伝子同士が近接する
また、ChIP-seqデータを比較するパッケージも存在しますが、注意して使用する必要があります。
典型的にはChIP-seqのバッチ効果は非常に大きく、異なるラボでのデータを比較することは簡単ではありません。実際、同じ研究室であっても昔のデータや昔のシーケンスマシンで得たデータと現在のChIP-seqのデータを単純に比較できないのです。
解析に起因するバイアスを除去するためにも、比較の際はパイプライン処理をもう一度生データから実施し直すことをお勧めいたします。

